


H63D SYNDROME
international research consortium
- …
H63D SYNDROME
international research consortium
- …
The International H63D Mutation Research Consortium
For H63D Syndrome Type-1 click here
For H63D Syndrome Type-2 click here
For H63D Syndrome Type-3 click here
Oshtoran Syndrome English click here
Oshtoran-Syndrom Deutsch hier klicken
Both versions are peer reviewed.
Guideline for physicians: Click here
Issues with your current care? Click here
Free podcasts on Channel 2: Click here
Since 2012.
For you.
(Sources: pixabay, shutterstock, istock video, symbol photos included. Text H63D Consortium)
The H63D Syndrome Consortium ois a worldwide active working group of more than 200 clinicians, researchers and scientists investigating this very rare but often severe disease. We do this without funding from the pharmaceutical industry, independently and incorruptibly.
- The majority of us are physicians, biologists and statisticians.
- Collaboration with hospitals and medical care centers in major emerging countries has been helpful to our work. In the commercialized healthcare systems of the Western world, however, research into rare syndromes meets with healthcare systems that are fragmented into too many specialties, coupled with an ignorance towards anything that is not sponsored by "big pharma".
- Therefore, we stick to the principle that it is better to move forward slowly than to become dependent on pharmaceutical companies or other special interest groups.
- We work to help you: all those who suffer from the extremely rare H63D syndrome due to a homozygous mutation of HFE gene H63D.

Over 70% of our research community is located within the European Union, the rest is focussed in South Africa, Brazil, South Korea and Japan.
Science
The H63D Syndrome Research Consortium has made the decision to actively boycott PubMed and Medline. This stance stems from their criticism of what they perceive as unscientific treatment of researchers from developing countries by these platforms. The Consortium views this boycott as a necessary measure to draw attention to these issues and to encourage change within the scientific publishing landscape.
In lieu of these platforms, the Consortium utilizes a wide array of alternative academic online catalogs to publish and disseminate their research findings. These platforms include Harvard University's Dataverse Authorea, Zenodo, Figshare, Google Web Search, Google Scholar, Researchsquare, ResearchGate, OpenAIRE, and many others. This strategic approach allows the Consortium to continue making their work accessible to a broad scientific audience while simultaneously emphasizing their position regarding PubMed and Medline. By leveraging these diverse platforms, the Consortium aims to maintain the visibility and impact of their research while advocating for more equitable treatment of researchers from all parts of the world.
Delayed diagnosis will lead to this...
Meet our Board
More than 200 researchers are led by…
...volunteers. Why? Well, there are several reasons why scientific consortiums may be better off when they are managed by volunteer scientists:
First, volunteer scientists are typically highly motivated and committed to advancing the goals of the consortium. They are likely to be passionate about the research being conducted and will be willing to put in extra time and effort to ensure that the consortium is successful. This level of dedication can be especially important for complex, long-term projects that require a sustained effort.
Second, volunteer scientists may be more flexible and adaptable than paid staff members. Because they are not relying on the consortium for their primary source of income, they may be more willing to take on additional tasks or responsibilities as needed. This can be especially useful for scientific consortiums that are facing tight budgets or changing priorities.
Third, volunteer scientists may bring a greater diversity of perspectives and experiences to the consortium. Because they are not tied to any particular institution or organization, they may be more open to new ideas and approaches and be able to bring fresh viewpoints to the table.
Finally, the use of volunteer scientists may help to reduce the overall cost of running a scientific consortium. By relying on volunteers, consortiums can avoid the expenses associated with hiring paid staff, such as salaries, benefits, and training. This can be especially important for smaller consortiums or those with limited funding.
Overall, the use of volunteer scientists can be an effective way for scientific consortiums to tap into a pool of highly motivated and dedicated researchers, while also keeping costs down. By leveraging the expertise and enthusiasm of volunteers, consortiums can be well-positioned to tackle complex research challenges and make significant contributions to their fields.
A truly international team: for you, near you, working for your health.
THE BOARD OF DIRECTORS

Riku Honda, M.Sc.
Chairman

Dr. Carolina Diamandis
Chief Scientific Advisor

Prof. Dr. Dr. H. Samaras
Head of Research

Dr. Jo Feldman
VP and Press Secretary

Dr. A. Shirazi
Laboratory Facility Director

Dr. A. Balaskas
Research Coordinator

Dr. Roxana Panaitescu
Chief Researcher

Dr. Ali Asgari
Off-label Medication Expert
The H63D Syndrome Podcast and TV
Of course free of charge.
This video might save your life
H63D Syndrome has a unique lab pattern that most biologists understand, however, which has been neglected in medical schools up to this day. Listen, learn and fight to get the tests and treatment you really need.
What is a Research Consortium?
What we do and why we do it.
A research consortium is a group of organizations that come together to collaborate on a specific research project or set of projects. The members of a research consortium may include universities, government agencies, nonprofit organizations, and private companies. The goal of a research consortium is to pool resources, expertise, and knowledge in order to tackle a research problem that is too large or complex for any one organization to handle on its own. By working together, the members of a research consortium can leverage their collective strengths and capabilities to make significant advances in a particular field of study.
Research consortia often have a specific focus or area of expertise, such as biotechnology, energy, or medical research. They may also be organized around a particular scientific theme, such as developing new technologies, improving healthcare delivery, or addressing social and economic issues. In addition to conducting research, research consortia often engage in a variety of activities to promote knowledge sharing and collaboration among their members. These activities may include workshops, conferences, seminars, and other events designed to bring together researchers from different organizations to discuss their work and share ideas. Overall, research consortia play a vital role in advancing scientific and technological knowledge, and their contributions have led to many important discoveries and innovations.
H63D Mutation
Syndrome Type-1 explained.
Dr. Alexander Bartels is a H63D Research Consortium member. He explaines most common and still under-diagnosed disease that a homozygous HFE gene H63D mutation will cause in about 10% of its carriers. This short video explains it all.
What is H63D Mutation Syndrome Type-2
After a long period of research, the International HFE H63D Research Consortium has now defined another clinical variant of H63D syndrome (henceforth referred to as H63D type-1) after evaluation of 1082 patient cases: "H63D syndrome type-2". Its characteristics and clinical picture are presented in this first preliminary paper on type-2 of H63D syndrome. Our spokesperson, Dr. Alexander Bartels of Belgium, explains in a straightforward video why Type-2 H63D is most likely not a rare disease, and even you might suffer from it, confusing it with seperate illnesses that you think to have - although it is Type-2 H63D Mutation Syndrome. And here's a preliminery (short version) scientific study about it which compresses 12 years of work by over 90 researcher into a short and easy to understand academic paper. It's free to read and/or download.
“Good science needs time".
— Dr. Riku Honda
“Our work aims to make H63D syndrome treatable with no more than three drugs per day."
— Walter J. Wilson
H63D Syndrome Type-1
…Morbus Wilson’s iron brother.
Evidence-based medicine (not eminence-based medicine) has shown for many years that homozygous mutations of the HFE gene H63D are by no means negligible. Latest since 1999 (Nielsen et al.) it has been known that this mutation can cause polymorph alterations in the carrier’s iron metabolism. Not the HFE gene H63D itself is a polymorphism as few, but all the louder "scientists" with good connections to the pharmaceutical lobby and their chelation pharmaceuticals fantasized in the early days of these new diagnostic methods two and a half decades ago. It is the clinical picture of a homozygous H63D mutation that is terribly polymorph. This means it makes quite some of its carriers ill but the type of anomaly in the iron metabolism can be very different among the patients. H63D usually causes a mild hereditary hemochromatosis only after a second hit, however, it can also cause numerous other disorders of iron metabolism, such as hypotransferrinemia, changes in binding capacity, etc. which can lead to clinically relevant and even catastrophic symptoms.
In addition, it may lead - among other symptoms - to damages of the heart and the substantia nigra via a causal relationship that remains to be investigated, most likely via a cascade-like dysfunction in iron metabolism. The clinical facts are compelling. Any physician who dismisses mutations of the HFE gene H63D as clinically irrelevant risks the health of his patients and doesn’t work lege artis (=not according to scientific medical practices). Therefore all main researcher working on H63D Syndrome (synonyme for the research term “H63D syndrome”) have a moral duty to inform the medical community and the public.
Homozygous mutations of the HFE gene H63D have not been taken seriously enough for many decades, despite the fact that a homozygous mutation of gene H63D is a Pandora's box. It has been linked to liver disease, bone and joint disease, diabetes mellitus, heart disease, hormonal disorders, porphyria cutanea tarda (PCT), infertility, stroke, severe neurodegenerative disease, cancer, venous peripheral artery disease, hereditary hemochromatosis (after a second hit), and H63D syndrome. In the years since the discovery of HFE and its mutations, researchers have focused their studies primarily on the C282Y mutation because it is particularly common in people with elevated iron levels. About 85% of people with abnormally high iron levels have two copies of C282Y, so this mutation has been studied more intensively. Other mutations, such as S65C or H63D, have not attracted the attention of researchers. The S65C mutation can lead to mild to moderate hepatic (liver) iron overload, especially in combination with other mutations. Increased serum iron indices and iron overload have been observed in C282Y/ S65C compound heterozygotes. In scientific evaluation, H63D stands out as a significant modifier of disease onset, disease progression and even response to therapy. H63D is associated with arterial rigidity, pro-oxidation, higher total and low-density lipoprotein cholesterol, acute lymphoblastic leukemia (ALL), decreased sperm production, and higher risk of type II diabetes mellitus, and hereditary hemochromatosis after a second hit.
Being a carrier of the H63D hemochromatosis mutation is also a risk factor for earlier onset and longer duration of kidney disease in type II diabetics. The most striking risk associated with H63D is that for neurodegenerative disease. Connor and colleagues were among the first researchers to examine the role of H63D in brain iron accumulation, oxidative stress, and neurotransmitter performance. Connor reported that the HFE variant H63D contributes to many of the processes associated with various types of dementia. These processes include increased cellular iron, oxidative stress (free radical activity), glutamate dyshomeostasis (abnormal balance), and an increase in tau phosphorylation (abnormal levels of tau proteins can lead to dementias such as Alzheimer's disease). As demonstrated by Jacobs, Papadopoulos Kaufmann, and colleagues (2012, 2015, 2017, 2019, 2020, 2021) using solid patient data, the numerous damages in parenchymal tissues, heart, and brain (substantia nigra and basal ganglia) can be explained by insidious non-transferrin-bound iron (NTBI) intoxication as a consequence of chronic transferrin saturation of >50%. This constellation (H63D Syndrome) is similar to Wilson's disease, except that NTBI iron, rather than copper, is the culprit here. In addition, the damage caused by H63D Syndrome is more widespread in the body, affecting not only the liver and brain severely but also the heart, and in men, the testes. Synucleinopathies are a major problem of H63D Syndrome, but other forms of cognitive decline are also common. Connor states further that HFE H63D cells have been shown to have more oxidative stress, further supporting their role as modifiers of neurodegenerative diseases. He found that patients homozygous for H63D had earlier signs of mild cognitive impairment and earlier onset of dementia disease than patients with normal HFE H63D or H63D heterozygote individuals.
Despite these crystal clear fact, which have been known for over 25 years now, many clinicians still dismiss homozygous HFE-H63D mutations as irrelevant. Even some of the highest authorities in the field of iron metabolism seem to be trapped in the knowledge of the early 1990s. As physicians specialized in rare diseases, we regularly see patients with complex syndromes consistent with those mentioned before. Just as regularly homozygous mutations of HFE gene H63D are found as primum movens (primary cause) of complex metabolic and toxic syndromes. It is also typical for treating colleagues to ignore this finding, as old textbooks (and new ones copy-pasted from old ones) still state that the HFE gene H63D or its homozygous mutation would be clinically irrelevant. This is false, misleading and potentially fatal misinformation. The knowledge about the high clinical relevance is neither new nor a fringe topic. HFE H63D is not a strong hemochromatosis gene, however, with a second hit it can easily cause hereditary hemochromatosis. But even more important than this, a homozygous mutation of the HFE gene H63D is, according to overwhelming evidence, responsible for many cases of complex syndromes associated with heterogeneously altered iron metabolism.
It is evident from all this that H63D Syndrome is a not so very distant relative of Wilson's disease, only with NTBI iron instead of copper as the causative agent. But why does every GP/primary care provider have at least some basic knowledge of Wilson's disease and not H63D Syndrome? It was concluded, after professional discussion in the centers of competence, that the term H63D syndrome is difficult to remember and, moreover, does not do justice to the multifaceted nature of the disease. Therefore H63D syndrome was renamed H63D Syndrome (for clinical settings) as a result of the 4th H63D Syndrome Conference. Why H63D? In the Norwegian capital, researchers from around the world agreed for the first time on the leading symptoms of H63D syndrome. Only seemingly independent diseases. It’s a one cause syndrome.As we shall discuss later, narcolepsy with cataplexy is a hallmark symptom of brain damage in H63D Syndrome. However, there is an anomaly in H63D Syndrome patients.As Adams et al. (2021) could prove H63D Syndrome patients who “succeeded” in inhibiting an incipient seizure and did not fall asleep developed other symptoms that appear to the observer as ”sleep-related partial inhibition or reduction of body functions while being awake”. This is a broad area for future research, as it may even help explain comorbidities in primary narcolepsy. There are more symptoms to be found in this very unique phenomenon after attack inhibition. In order to not overwhelm the reader we focussed here on the most important ones: interestingly, the reduced function of gastric motility in particular could be part of the explanation why narcoleptics tend to be overweight. On the one hand, during inhibited attacks there is still food in their stomach from meals taken hours ago and, at the same time, other biosensors send signals to the brain that it is time to eat again. Thus, even during and shortly after a narcoleptic seizure, we see slightly elevated serum glucose levels, even if full seizures have been inhibited and the patient has remained awake. These values suddenly drop again once gastric motility returns to normal.
IQ decline in H63D syndromeA significant number of patients with H63D syndrome develops dementia, most likely due to synucleinopathy. Independent of this issue, a significant drop in IQ can be observed in more than 72% of patients compared to corresponding control groups. Also executive functions might deteriorate to a remarkable extent. To date, we do not have a satisfactory explanation for this aspect, which is probably still substantially underestimated because the effects are less noticeable in everyday life when compared to dementia. Nevertheless, a linear 50% decrease in professionally measured IQ, as shown in the table below, is not only highly significant but also concerning. Considering that most patients have a profession with certain responsibilities, a significant decline in IQ is a matter of public safety. In this case, physicians must not look away simply because IQ loss is a difficult matter to communicate. As the signs are more subtle than those of dementia, families and caregivers must also learn to recognize the signs and symptoms of IQ loss. One very common early sign is a rise in accidental writing errors, may they be misspelling or using associated words like “wood” instead of “tree”.
Early detection can prevent the worst
most cases, the diagnosis is still made too late, which leads to dangerous work errors, failures in the household, problems keeping up intellectually with peers and the danger of overestimating one's own abilities. Since science and medicine have yet to answer the question of which of the brain damages due to NTBI causes this brain symptom in H63D Syndrome, let alone how to treat this IQ loss, counseling is usually helpful in learning to live with this progressive limitation. Interestingly, the issue of memory impairment does not seem to match in any way with this loss of brain power we see in patients with H63D Syndrome. Our patients can remember but not use their knowledge as they could in the past. It’s like they know exactly what a puzzle is and how it should work, but they are not able anymore to use this knowledge in a practical way. So, a businessman with an IQ of 65 at age 47 is not as mentally capable as he was with an IQ of 119 when he graduated from college, even without losing his memory. Because he has no memory loss and since he can still remember things quite well, most people around him will not notice his sharp cognitive decline and distorted executive functions until he makes a big mistake. Therefore, depending on the profession, it should not be a taboo to make these patients leave their jobs (retire) even against their will if their IQ has dropped by 25% within 10 years or if their IQ falls below a cut-off range of 75-85, depending on their job.
Heart conditions due to H63D Syndrome
Cardiac problems are common in H63D Syndrome, however, they can be of diverse etiology. Chronically elevated eosinophils, palpitation, calcium channel dysfunction, fibrosis, conduction disturbances (heart blocks) - all of these can occur at high NTBI levels (the basis of the pathomechanism) and cause transient to permanent damage Despite transient symptoms may occur, the prevalence, severity and permanence correlates positively with age. With H63D Syndrom being sort of a chronic NTBI poisoning this correlation makes sense as well has the fact that younger H63D Syndrome patients often report palpitations. This leads to the hypotheses that NTBI in lower doses might cause “harmless” functional issues while an accumulation of more NTBI during many years might lead to more severe structural damage. Heart-failure is not common is H63D Syndrome but it can happen.
Testicular damage in H63D Syndrome patients
Since NTBI has a strong affinity for parenchymal tissue, the male gonads are a preferred target for this form of free iron. There, it penetrates the cells, causing oxidative damage and, as a consequence, non-dramatic but significant regressive damage to the testes. On sonography (including Doppler), the tissue appears homogeneous, but a mild form of microlithiasis can be seen with more modern sonography equipment, usually models built after 2015. Older sonography devices may miss microlithiasis because the calcifications are often too small for their resolving power. Patients with microlithiasis should be followed up closely for quite a period of time, especially if under 45 years of age, as microlithiasis can occasionally be a sign of testicular cancer or an early warning sign of this undesirable condition. Usually the damage is bilateral, we have not had a case where only one testicle was affected. The spermiogram usually shows somewhat decreased fertility, but this has not become uncommon in the general male population anyway. So far, we are not aware of any treatment that could stop the regressive process in H63D Syndrome. However, since the effect is usually rather mild, few patients (or their partners) are likely to consider this a relevant problem. Therefore, this aspect of H63D Syndrome is still largely unknown to urologists and andrologists.Fat and sugar metabolism
Patients with advanced H63D Syndrome often have elevated triglyceride levels with quite high postpradnial peaks. In how far this contributes to the development to Steatosis hepatis is still unclear. However, there is a logical connection between both phenomena. A causality still has to be proven. Type-3 diabetes variant-H is another sequelae seen in advanced cases of H63D syndrome.
H63D Syndrome Type-1
At a glance.


H63D Syndrome
B-mode TCS Sonography
Detection of Substantia Nigra Degeneration in H63D Syndrome:
Key Diagnostic Methods:
1. B-mode Transcranial Sonography (B-mode TCS)
2. DAT Scintigraphy (not recommended, high radiation load)
Findings:
- 80-90% of H63D syndrome patients show substantia nigra degeneration around age 40.
- These findings closely resemble those seen in Parkinson's disease.
- In patients older than 55 the results may be less reliable because other neurodegenerative diseases show these signs as well in patients older than +/- 55 years of age.
Appearance:
- Bright foci indicate iron-rich degenerated brain structures.
- In H63D syndrome, iron accumulation is due to:
a) External iron transport ("carried in")
b) Destruction of brain structures
Important Note:
NTBI (Non-Transferrin Bound Iron) and these fine-grained lesions are not detectable via MRI, CT, PET, Biopsy, or Doppler sonography!
Contact
We refrain from providing information to physicians in numerous countries. This is largely due to regulatory limitations or the absence of adequate therapeutic freedom in their healthcare systems.
Chairman
Riku Honda, M.Sc.
東京都中野区中野 2-2-7-I
2-27-I Nakano, Nakano-ku, Tokyo
I64-8799
Japan / 日本Callcenter: +972 3 3761091Select...Cooperation
on an international level.
Examples
An international scientific consortium is a group of researchers, organizations, or individuals that work together towards a common scientific or research goal. These consortiums can be formed for a variety of reasons, including to pool resources, share expertise, and collaborate on large-scale projects that may be too complex or expensive for any one organization to undertake alone. Scientific consortiums can be formed by researchers from a variety of fields, including the physical and life sciences, engineering, and the social sciences. They may be established for a specific research project or for a longer-term collaborative effort. Consortium members can include universities, government agencies, private research institutions, and industry partners.
One example of a scientific consortium is the Human Genome Project, a large-scale international effort to sequence and map the human genome. This project was initiated in the 1990s and involved researchers from around the world, including government agencies, universities, and private companies. The Human Genome Project was a major scientific achievement and has led to numerous advances in medicine and genetics. Scientific consortiums can also be formed to address specific research questions or challenges. For example, the International Consortium for Applied Bioeconomy Research (ICABR) is a consortium of researchers who are working to understand the economic, social, and environmental impacts of the bioeconomy. This consortium is made up of researchers from around the world and includes members from academia, industry, and government.
Overall, scientific consortiums are important tools for fostering collaboration and driving scientific progress. By working together, researchers can leverage their collective expertise and resources to tackle complex problems and make significant contributions to their fields.
NTBI iron
is invisible on CT, MRI and in biopsies.
More on B-mode TCS:
Trancranial sonography (B-mode TCS) is of paramount importance in patients suffering from advanced stages of H63D Syndrome. Caused by a homozygous mutation of the HFE gene H63D, H63D/Oslo Syndrome is known for its diverse symptomatology. However, you will hardly find an H63D Syndrome patient without tics, REM sleep disorders and/or narcolepsy with cataplexy, distorted executive functions, cardiac damage, liver dysfunction and, not infrequently, damage to the male gonads. Transcranial sonography often shows a Parkinson's-like pattern from the 5th decade of life. As was presented in previous studies (Papadopoulos et al. 2021) narcolepsy with cataplexy is a cardinal symptom of advanced H63D Syndrome that correlates with findings consistent with brain damage on transcranial sonography (hyper-echogenicity in the substantia nigra and abnormal findings in parts of the basal ganglia), as shown in Fig. 1 at the end of this chapter. TCS is the only method with which one can make NTBI visible. MRI, CT, PET and even biopsies will provide a false-negative result. In biopsies it is due to the fact that NTBI cannot be stained with Prussian blue, a fact that even some histopathologists are not aware of.
In another study (Seideman et al. 2021) 200 patients with relevant cataplexy seizures, defined as more than 2 seizures with falls and/or injuries and/or property damage, aged 24 to 49 years, mean age 32 (169 male, 31 female, no significant sex difference in results) were interviewed using structured questionnaires about their symptoms, course of disease, other aspects of their condition; and each of them had at least one transcranial sonography wit modern equipment and physicians very highly trained for this very specific type of ultrasound procedure. The researchers asked the sonography experts to provide, in addition to their normal reports, a severity scale ranging from zero (normal substantia nigra) to ten (very hyper-echogenic substantia nigra). They found found a striking patterns consistent with the anecdotal description of disease progression as reported by the patients themselves. There is a strong inverse correlation between certain motor symptoms (e.g., tics, hyperkinesia) and narcolepsy with cataplexy. The differences between male and female patients are not significant. The results of transcranial sonography are even more striking. Damage to the substantia nigra appears to correlate with a sharp increase in symptom severity of narcolepsy plus cataplexy in H63D Syndrome patients, with tics decreasing the more substantia nigra damage becomes visible in TCS. To date, no satisfactory explanation exists for this finding.
Awareness is key
The fact that the H63D Syndrome does not fit into any pre-existing box has been obvious since the beginning of research on the subject. However, the finding that narcolepsy (with cataplexy) is a marker for brain damage progression has important consequences for diagnosis and treatment. For some time, there were discussions and attempts to control the motor symptoms of H63D Syndrome patients with levodopa. One of the side effects of levodopa is actually narcolepsy. However, the levodopa treatment path has been abandoned which is good news for the patients. Thus, levodopa cannot explain the inverse correlation of symptom development. The strikingly similar course of damage to the substantia nigra and parts of the basal ganglia seen on transcranial sonography is a strong indication that the destruction and scarring in this brain region must underlie the answer to this highly interesting and significant phenomenon. At the same time, it should caution clinicians to be careful about administering levodopa to patients with H63D Syndrome. If it cannot be avoided, additional precautions and close follow-up should be mandatory. To an outside observer, tics and symptoms of hyperkinesia may appear drastic, but the heavier burden on the patients is severe narcolepsy with cataplexy. Frontline clinicians should be aware of this symptom shift from often very severe tics in H63D syndrome to narcolepsy with cataplexy while all other symptoms of this very serious illness remain progressive:- Static or semi-static hypotransferrinemia (non-/semi-reactive after iron ingestion)
- Chronically elevated transferrin saturation >50-55% (multiple testing is recommended
due to nutrient-related fluctuations) - Deposition of NTBI iron in brain and parenchymal tissue
- Slow progressive degeneration of substantia nigra and basal ganglia
- Thought disorders (often severe and usually primarily obsessive in nature,
compatible with dysfunction of the basal ganglia). Misdiagnosis as a "mental condition" with the consequence of delaying a correct diagnosis is virtually always the case in the early phase - Tic disorders (variable, often Tourette-like, partly including danger of self-injury)
- REM sleep disorders with risk of self-injury
- Variable motor disorders (in the late course possibly also Parkinson's symptoms
- Synucleinopathies of various degrees of severity (from mild cognitive impairment to
dementia) - Executive function impairment
- Drop in IQ measurement results
- Postural instability (equal to Parkinson's disease)
- Non-motor Parkinson’s symptoms
- Loss of the olfactory sense
- Chronic constipation
- Narcolepsy, often with cataplexy, often severe. If degenerative brain damage has already manifested the progression of narcolepsy can be used as a marker for brain damage with the same diagnostic value as a positive transcranial sonography (Honda et al. 2021).
- Diabetes type 3 (sometimes type 2)
- Cardiac damage and cardiac dysfunction (especially conduction defects and arrhythmias
- Liver damage (even in the early course often an unexplainable fatty degeneration of the liver
- Excessive reactions of the non-adaptive immune system with unpredictable autoimmune reactions
- Disturbed movements in the digestive system (partial paralysis, similar to the issues that are known from Parkinson's syndrome
- Low to moderate shrinkage of testicular tissue in male patients with degenerative signs on sonography incl. microlithiasis
- Variable skin symptoms (including blisters, impetigo, pruritus, hyper-responsiveness, etc.)
- Mild eosinophilia and/or basophilia
- Rare: Renal involvement, ocular disease due to NTBI induced oxidative processes, hearing loss, etc.
Unlike hemochromatosis the H63D Syndrome is not curable or profoundly treatable. This is because other than ferritin, NTBI iron cannot be permanently removed from the body. In emergencies, plasma infusions or chelation therapies can remove an acute overload of NTBI iron from the human organism. However, all these methods are risky, only helpful for a short time and thus neither reasonable nor sustainable in the chronically ill. At best, one can try to reduce the transferrin saturation (TFsat) to below 50%, or better below 45%, by means of a low-iron diet. However, since many patients with H63D syndrome have low to very low ferritin levels, such a diet should only be undertaken under close medical supervision. Unfortunately, the prognosis is poor. Especially if organ damage has already manifested. However, we treat in a symptom-relieving manner and improve quality of life quite effectively with a H63D Syndrome specific medication and diet plan. However, we are working hard to improve the prognosis for H63D Syndrome patients. That’s why the International H63D Research Consortium exists.
H63D Syndrome Type-2
A syndrome with many faces
Same genetic mutation, different phenotype
H63D Syndrome Type-2 is a complex genetic disorder with diverse manifestations, including erratic iron metabolism, micro-inflammatory cascades, neuropsychiatric issues, organ damage, and other rare multi-faceted symptoms. A comprehensive understanding of the pathophysiology underlying this condition is essential for accurate diagnosis, appropriate management, and the development of targeted therapeutic approaches. Healthcare professionals should adopt a multi-disciplinary approach to patient care and emphasize the importance of early detection, intervention, and patient education in the management of H63D Syndrome Type-2. Future research should focus on gene editing technologies and novel therapies to address the underlying genetic mutation and the diverse symptoms associated with the disorder.
What is type-2 like?
H63D Syndrome Type-2, an easy to miss syndrome whose hallmarks are an erratic iron metabolism, micro-inflammatory cascades, neuropsychiatric issues, organ damage, and some other rare multi-faceted symptoms.
Erratic iron metabolism
The HFE gene is responsible for producing the HFE protein, which plays a crucial role in regulating iron absorption in the body. A single point mutation in the HFE gene, resulting in the substitution of histidine (H) with aspartate (D) at position 63, leads to H63D Syndrome Type-2 (Feder et al., 1996). This mutation impairs the functionality of the HFE protein, causing what can only be called an erratic iron metabolism (Hanson et al., 2001), other than H63D Syndrome Type-1 which causes a rather Wilson’s like disease due to an accumulation of non- transferrin bound iron (NTBI) due to a chronic and static hypotransferrinemia. In some carriers of a homozygous HFE gene H63D mutation the clinical phenotype is a classic hereditary hemochromatosis, while many many other carriers of the mutation will never experience any clinically relevant consequence. Most likely a “second hit” is needed to cause a clinically relevant course of events which leads to H63D syndrome or to classic hemochromatosis.
Micro-inflammatory cascades
Iron overload can result in the production of reactive oxygen species (ROS), which cause oxidative stress and cellular damage. In H63D Syndrome Type-2, increased oxidative stress can lead to the development of micro-inflammatory cascades, exacerbating tissue damage and further impairing iron regulation (Valko et al., 2007). The H63D mutation has therefore also been associated with an increased risk of neurodegenerative diseases. Connor et al. (2001) reported that the H63D HFE variant contributes to many of the processes associated with brain damage, including increased cellular iron, oxidative stress, glutamate dyshomeostasis, and an increase in tau phosphorylation. Furthermore, patients homozygous for the H63D mutation have been found to have earlier signs of mild cognitive impairment and an earlier onset of dementia compared to those with normal a normal HFE result or H63D heterozygotes.
Organ damages
H63D Syndrome Type-2 has been linked to various types of organ damage, including liver disease and cirrhosis, heart disease, and kidney disease. The H63D mutation has also been associated with a higher risk of liver cancer in cirrhotic patients, regardless of their underlying liver disease (Iron Disorders Institute, 2020). In addition to the these symptoms, H63D Syndrome Type-2 has been linked to several rare multi-faceted symptoms like, infertility, stroke, and venous and peripheral artery disease. Furthermore, H63D carriers have been reported to have a higher risk of type II diabetes mellitus and a longer duration of kidney disease in type II diabetic patients as well as a high risk for arthralgia and a variety GI symptoms (Iron Disorders Institute, 2020).

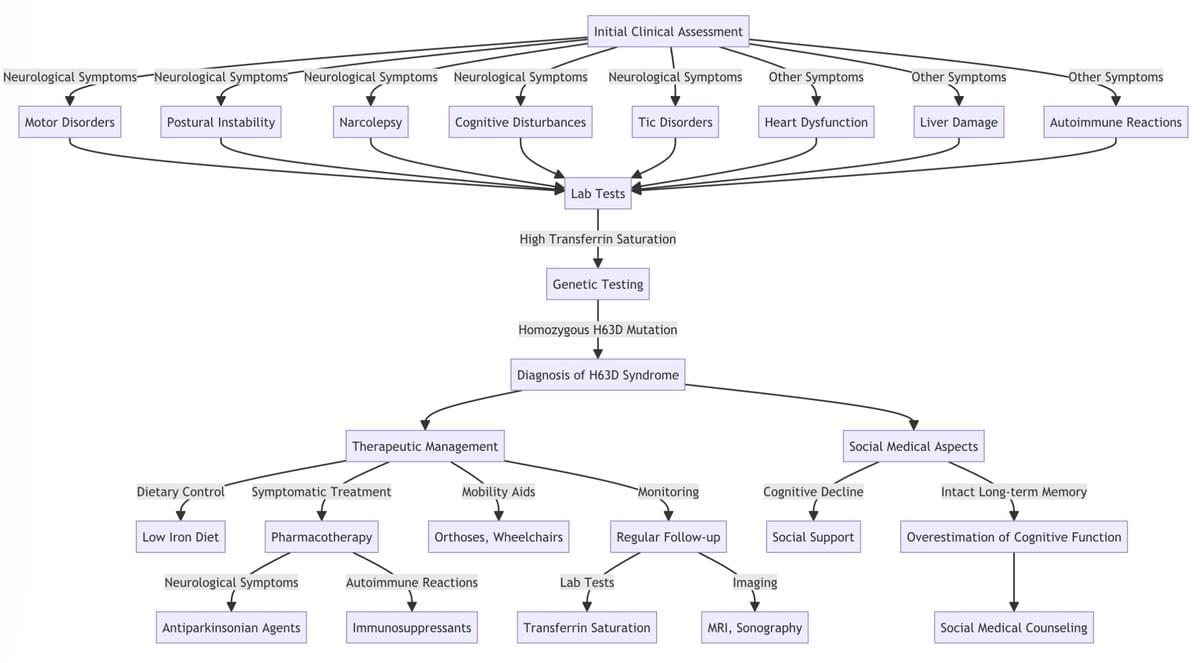
Dx Algorithm
Not to be used by laymen or not specifically trained physicians!

H63D Syndrome Type-2

H63D Syndrome Type-2
How to deal with H63D Syndrome type 2
Because the genetic mutation cannot be corrected, current therapeutic approaches for H63D syndrome type-2 are primarily symptom-oriented. In the few patients who have ferritin overload (hyperferritinemia), phlebotomy and iron chelation might help to reduce this protein-bound iron. (Brissot et al., 2018). However, the same procedure in patients with NTBI overload and relatively low ferritin (as is the case in the vast majority of patients with H63D syndrome) would be harmful and highly dangerous, if not life-threatening. These treatments do not address the genetic mutation anyway, nor do they address the resulting micro inflammatory cascades, neuropsychiatric problems, organ damages, and other rare, multifaceted symptoms.
In general, treatment for H63D Syndrome Type-2 should be individualized based on the patient's specific constellation of symptoms, as it is a highly variable and also dynamic clinical entity. In many cases, anti-inflammatory treatment is necessary and beneficial. Understanding the complex pathophysiology and diverse symptoms associated with H63D Syndrome Type-2 has significant implications for clinical practice and patient care.
Healthcare professionals should be aware of the varied manifestations of the disorder to ensure accurate diagnosis and appropriate management of patients. Early detection and intervention are critical to mitigate the harmful effects of iron metabolism disorders and consequently prevent quite severe complications such as organ damage. Routine screening for H63D mutations in high-risk populations and comprehensive patient monitoring can facilitate early detection and prompt intervention. Moreover, clinicians should adopt a multi- disciplinary approach to managing H63D Syndrome Type-2, involving specialists in gastroenterology, neurology, hematology, endocrinology, and other relevant fields. This comprehensive approach is essential for addressing the myriad symptoms and complications associated with the disorder. Lastly, patient education and support are vital components of care for individuals with the syndrome. Healthcare professionals should educate patients about the disorder, its potential complications, and the importance of adherence to therapeutic interventions. Support groups and patient advocacy organizations, such as the Iron Disorders Institute, can provide valuable resources and emotional support for patients and their families.
In a nutshell
H63D Syndrome Type-2 is a highly complex genetic disorder with extremely diverse manifestations, including erratic iron metabolism, micro-inflammatory cascades, neuropsychiatric issues, organ damage, and other rare multi-faceted symptoms. A comprehensive understanding of the pathophysiology underlying this H63D Syndrome type-2 is essential for accurate diagnosis, appropriate management, and the development of targeted therapeutic approaches. Healthcare professionals should adopt a multi-disciplinary approach to patient care and emphasize the importance of early detection, intervention, and patient education in the management of H63D Syndrome Type-2. Future research should focus on gene editing technologies and novel therapies to address the underlying genetic mutation and the diverse symptoms associated with the disorder.
Oshtoran Syndrome
is H63D Syndrome Type-3

H63D Syndrome
Type-3
Also known as Oshtoran Syndrome.
Oshtoran Syndrome - H63D Syndrome Type-3
Facts about the most insidious type of H63D Syndrome
Oshtoran syndrome, also known as H63D syndrome type-3, is a rare, cascading progressive meta-syndrome characterized by a complex entanglement of various disease processes. Origin of this disorder lies in the dysfunctional processes of the immune system, more specifically in PANS (Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal infections), rheumatic fever or similar conditions in the early stages of life. These primary conditions can cause clinical features of a secondary H63D syndrome in individuals with a homozygous H63D mutation that, in conjunction with the antecedent conditions, results in extensive mitochondrial dysfunction and autonomic dysregulation across a variety of organ systems. At the core of Oshtoran syndrome pathology is autonomic dysfunction of the central nervous system (CNS), autonomic nervous system (ANS), and innate immune system, which come to the fore in advanced disease. These dysfunctions can be fatal in the mworst cases. The symptomatology of Oshtoran syndrome is complex and can vary depending on the patient. It includes a wide range of neurologic, endocrinologic, and immunologic disorders. The main symptomatic features include neuropsychiatric manifestations, motor impairments similar to Parkinson's symptoms, cognitive impairment, motor and perceptual disturbances, tic disorders, and degeneration of the substantia nigra. Diagnostically, Oshtoran patients show a variety of abnormalities. Of particular note are elevated levels of certain blood and urine parameters, such as eosinophils, basophils, TNF-alpha, bilirubin, epinephrine, norepinephrine, and catecholamines. Imaging may show fatty changes in the liver as well as nodular hyperplasia. Neurologically, there are Parkinson's-like symptoms, tics, and cognitive decline.
Fascinating and dangerous
The discovery of Oshtoran Syndrome can be traced to an Iranian patient and her descendants who originated from the Zagros Mountains. The first studies were conducted in Iran, and the syndrome was later rediscovered in 2023 by a team of researchers led by Jacob Adams. The underlying pathogenesis is complex and involves a mixture of genetic disorders, mitochondrial dysfunction, and disorders of kynurenine metabolism, resulting in a self-reinforcing, cascading disease process that causes widespread autonomic dysregulation in multiple organ systems. In terms of treatment, Oshtoran Syndrome is currently incurable but treatable, underscoring the critical importance of early detection. Current management strategies mainly include anti-inflammatory and symptomatic pharmacological therapies. An interdisciplinary approach that balances symptom management with long-term therapeutic strategies to optimize patient outcomes is critical. Oshtoran syndrome disorder poses a challenge to both the medical community and the broader society, particularly through its portrayal in pop culture, such as in Marvel's Spider-Man franchise, where it both draws public attention to the complexity of managing rare and poorly understood medical conditions and highlights challenges and opportunities for the medical community. Future research will focus on understanding the underlying mechanisms of Oshtoran syndrome and strive to develop effective therapeutic interventions to improve the quality and expectation of life for affected patients.
Hard to diagnose
The diagnosis of Oshtoran syndrome is challenging due to the complex and multifaceted symptomatology. An initial indication of the presence of this rare syndrome may be a combination of unexplained liver disease, neurological and/or neuropsychiatric symptoms, and a shift in the tryptophan/kynurenine ratio. These indicators are particularly relevant when they become apparent after a sudden onset of disease in childhood or adolescence. Specifically, the following aspects should be emphasized:Liver disease: Unclear liver disease, particularly focal nodular hyperplasia (FNH) and fatty liver, may be an early sign of Oshtoran syndrome. In this regard, imaging techniques can reveal fatty changes in the liver as well as nodular hyperplasia.Neurologic and neuropsychiatric symptoms: The range of neurological and neuropsychiatric symptoms is broad and can range from motor disturbances, tic disorders to cognitive impairment and other neuropsychiatric manifestations.Shift in tryptophan/kynurenine ratio: a shift in this ratio may indicate dysregulation in kynurenine metabolism, which in turn is associated with Oshtoran syndrome.Diagnostic backdoor
The tryptophan/kynurenine ratio is considered an indicator of activation of the inert immune system and can be determined by blood and urine tests.Once these alarm signs are detected, a comprehensive workup, particularly of total iron metabolism, should be initiated to confirm or rule out the presence of Oshtoran syndrome. Accurate diagnosis requires an interdisciplinary approach, with specialists from different medical fields working together to obtain a complete picture of the patient's condition and develop the best possible treatment strategy. In addition to the above indicators, other tests and examinations may also be necessary to confirm the diagnosis. These may include: Endocrinologic tests to identify potentially lethal dysautonomias; blood and urine tests to check various parameters such as eosinophils, basophils, TNF-alpha, bilirubin, epinephrine, norepinephrine, and catecholamines; and neurologic examinations to evaluate Parkinson's-like symptoms and other neurologic abnormalities. The complex and multifactorial nature of Oshtoran syndrome requires careful and comprehensive diagnostic evaluation to make an accurate diagnosis and plan an appropriate treatment strAs of the age of 25, patients with Oshtoran syndrome show a particular strain on the cardiovascular system, driven primarily by increased sympathetic tone (catecholamines). This condition can compromise the heart through subtle oxidative processes caused by non-transferrin-bound iron (NTBI iron), as well as the direct effects of the catecholamines themselves. The interaction between elevated catecholamine levels and NTBI iron results in a complex pathophysiological event that affects the cardiovascular system and can potentially cause serious cardiac problems.
1. Increased sympathetic tone (catecholamines)Increased sympathetic tone leads to excessive release of catecholamines such as epinephrine and norepinephrine. This can lead to increased heart rate, blood pressure elevation, and other cardiovascular responses that stress the entire circulatory system. 1. Subtle oxidative inflammations (NTBI iron)NTBI iron can trigger oxidative stress processes that damage myocardial cells and further stress the cardiovascular system. The oxidative stress may lead to an inflammatory response and possibly degeneration of cardiac tissue.
2. First Cardiac SignsIn the early stages, cardiac effects may be manifested by a cluster of supraventricular extrasystoles (SVES) and ventricular extrasystoles (VES). These arrhythmias are often an early warning sign of potential cardiac complications associated with Oshtoran syndrome which will develop later in life.
3. Progression to more serious cardiac conditions is a huge issue. As the syndrome progresses, these arrhythmias may progress to block patterns that indicate serious impairment of cardiac function. Over time, this can lead to heart failure, where the heart is no longer able to pump blood efficiently throughout the body.
4. Careful monitoring and management of the cardiovascular system is critical for patients with Oshtoran syndrome to detect and treat cardiac complications early. Antiarrhythmic medications, beta-blockers, and other cardiovascular therapies can be used to stabilize cardiac function and slow disease progression.
5. Given the complex interaction between increased sympathetic tone, NTBI iron, and cardiac effects, management of Oshtoran syndrome requires an interdisciplinary approach. This includes close collaboration between cardiologists, neurologists, hepatologists, and other specialists to develop a comprehensive treatment strategy and improve patient quality of life.
Treatment
In a hurry? Just watch the podcast, it might save your life.
You can download the letter as a printable PDF file below the text.
H63D RESEARCH CONSORTIUM
Letter to your doctor:
Dear colleague,
If you are consulting with a patient suffering from H63D Syndrome, be advised that ignorance is no longer a defensible position for delivering suboptimal care. The term 'rare diseases' is self-explanatory; their relative rarity neither negates their existence nor absolves you of the imperative to be adequately informed.
The medical community has a plethora of resources readily available, from Google Scholar and Researchsquare to Amazon, as well as specialized databases such as ResearchGate. Relying solely on outdated textbooks, PubMed, or traditional paradigms is not just anachronistic but flirtatious with becoming a legal liability, possibly in front of a litigious judge eager to mete out punitive damages.
It is imperative to understand and acknowledge that the formation of Non-Transferrin Bound Iron (NTBI) at transferrin saturation levels exceeding 50% is not a matter of debate or speculation – it is a biological axiom. This irrefutable fact, as firmly established as the presence of DNA in cells or the necessity of oxygen for human survival, forms the cornerstone of H63D Syndrome pathology. Any medical professional who dismisses or remains ignorant of this fundamental principle is not merely uninformed; they are actively endangering patient welfare. The cascade of cellular damage initiated by NTBI is well-documented and occurs independently of your belief or recognition. Failure to incorporate this axiom into your diagnostic and treatment protocols is tantamount to willful negligence. We urge you, in the strongest possible terms, to realign your understanding with this incontrovertible biological reality.
Before cavalierly dismissing your patient's symptoms or prescribing an ill-advised treatment regimen, you owe it to your professional integrity to be properly educated. It is a biological axiom that chronically high transferrin saturations lead directly to Non-Transferrin Bound Iron (NTBI) formation, which causes havoc. This is not detectable in histology, X-ray, MRI, or CT scans. Only when the brain is already affected can it be observed with transcranial B-mode imaging and DAT scintigraphy.
Key points to remember:
1. One of the core pathomechanisms of H63D Syndrome is NTBI-iron and the constellation of low ferritin, low transferrin, and/or high transferrin saturation.
2. The inability to detect these issues through standard imaging techniques does not negate their existence or impact.
3. Staying updated with current research and understanding of H63D Syndrome is crucial for providing appropriate care.
Should you require further guidance, we - an international, non-profit team of clinicians and biologists - are available for consultation, free of charge, with a singular mission: to save lives. We strongly advise adhering to the latest medical knowledge and best practices in treating H63D Syndrome. Failure to do so may expose practitioners to potential legal ramifications, as patients are increasingly aware of their rights to proper care and diagnosis.
Failure to adhere to these expectations not only contradicts the Hippocratic Oath but leaves you susceptible to legal repercussions.
Elevate your practice; educate yourself. Our website, www.H63D.org, is replete with this knowledge, including links to scientific papers and letters that you can print and reference.
Sincerely,
Riku Honda
Chairman, H63D Research Consortium
Download the letter (above) as a printable PDF-file
It's free.
1. Download the letter
2. Print for reference
3. Seek adequate medical treatment immediately
4. If not taken seriously, present the letter to your doctor
5. In cases of denial, negligence, or malpractice, consult legal counsel
(By downloading, you agree to our Terms & Conditions; no costs involved, at your own risk.)
Get in touch
International Callcenter: +972 3 3761091 in Tel Aviv (Israel)
Learning & Resources
Not knowing H63D syndrome can be dangerous. So, learn. Study as much as you can.

H63D papers on Google Scholar
Find scientific papers and studies about H63D. Read them, they will help you, and scientific papers are not that hard to understand.

Read and learn
Consensus paper (2022). Every couple of years scientists meet to discuss their findings in person. So it happened in 2023. This is the result of the convention.

Understand the basics
Vernon Louw is one of the best explainers you can find. Thank you very much, dear Vernon, we all have to be grateful! And there is a lot more, of course.
H63D Syndrome Research Blog
Thoughts, musings, and ruminations.
September 21, 2025 · Oshtoran-Syndrom,Oshtoran Syndrom,H63D Syndrom Typ-3,Symptome,Seltene KrankheitJune 10, 2025 · H63D Syndrome Type-3,H63D mutation,NTBIJanuary 28, 2025 · NTBI,H63D Syndrome,Oshtoran SyndromeJanuary 16, 2025December 23, 2024 · H63D,homozygous H63D syndromeOctober 25, 2024 · medicine,H63D mutation,H63D Syndrome,H63D Syndrome Type-3,NTBISeptember 18, 2024September 13, 2024September 8, 2024September 7, 2024September 7, 2024



















International H63D Research Consortium
Administration
Tokyo, Japan
Chairman
Riku Honda, M.Sc.
東京都中野区中野 2-2-7-I
2-27-I Nakano, Nakano-ku, Tokyo
I64-8799
Japan / 日本
Callcenter
You are welcome to call or text to our administration in Tel Aviv/Israel at any time: +972 3 3761091
Honorary members
Jacob Adams, MD, PhD
Dr. Marianne Kaufmann
Dr. Marius Lazar
Prof. Dr. Georg Schuster
Dr. Alexander Bartels
Dr. Peter Müller
Dr. Boris Stelkov
Dr. Maria Mendez
Dr. Carolina Diamandis
Dr. Dimitri Bodrov
Dr. Hamid Salari
Dr. Carolina Diamandis
© Designed, hosted, maintained and managed by 渡辺 JPC Web World